Liquid structure research group

Department
Complex Fluid Research
Group Leader: Pál Jóvári
Understanding disordered structures. — Our research group is involved in the investigation
of short-range order of liquids, amorphous materials and disordered crystals. We combine
experimental data such as X-ray and neutron diffraction structure factors and EXAFS spectra
with computer modeling tools such as Reverse Monte Carlo (RMC) and molecular dynamics
(MD) simulations. As a result of such an approach, large configurations (typically tens of
thousands of atoms) are provided that are energetically reasonable and consistent (within
errors) with experimental data. These configurations are then subjected to various
geometrical analyses, so that specific questions concerning the structure of a material may
be answered. The group is also responsible for the maintenance and operation of the MTEST
neutron diffractometer installed at the 10 MW Budapest Research Reactor. Below, we
provide some selected results from the year of 2017.
Metallic glasses. — The structure of glassy Cu47.5Zr47.5Ag5 has been investigated by neutron
diffraction with isotopic substitution, X-ray diffraction as well as with Cu and Ag K-edge
extended X-ray absorption spectroscopy (EXAFS) measurements. Experimental datasets
have been fitted simultaneously with the reverse Monte Carlo simulation technique.
Nearest-neighbor distances and coordination numbers have been determined and
compared with those of glassy Cu50Zr50 and Cu47.5Zr47.5Al5. It has been found that the Cu-Cu
coordination number drops upon adding Al or Ag to Cu50Zr50. Both Ag and Al prefers Zr to
Cu. The total coordination number of Ag is 13.9±0.6 while that of Al is 10.2±1.0, suggesting
that, in spite of their similar molar volumes, the effective size of Ag and Al in the Cu-Zr
matrix is quite different. This is reflected both by the comparison of Zr-Al and Zr-Ag partial
pair distribution functions and the Zr-X-Zr (X=Al, Ag) cosine distributions (see Fig. 1.)
Alcohol-water mixtures. — Our efforts concerning the structure of alcohol-water liquid
mixtures have been extended to the study of the temperature dependence of the structure
of aqueous solutions of methanol. The evolution of the structure of liquid water-methanol
mixtures as a function of temperature has been studied by molecular dynamics simulations,
with a focus on hydrogen bonding. The combination of the OPLS-AA (all atom) potential
model of methanol and the widely used SPC/E water model has provided excellent
agreement with measured X-ray diffraction data over the temperature range between 298
and 213 K, for mixtures with methanol molar fractions of 0.2, 0.3 and 0.4. Hydrogen bonds
have been identified via a combined geometric/energetic, as well as via a purely geometric
definition. The number of recognizable hydrogen bonded ring structures in some cases
doubles while lowering the temperature from 298 to 213 K; the number of sixfold rings
increases most significantly. An evolution towards the structure of hexagonal ice, that
contains only sixfold hydrogen bonded rings, has thus been detected on cooling watermethanol
mixtures. For a picture of typical hydrogen-bonded ring structures, see Figure 2.
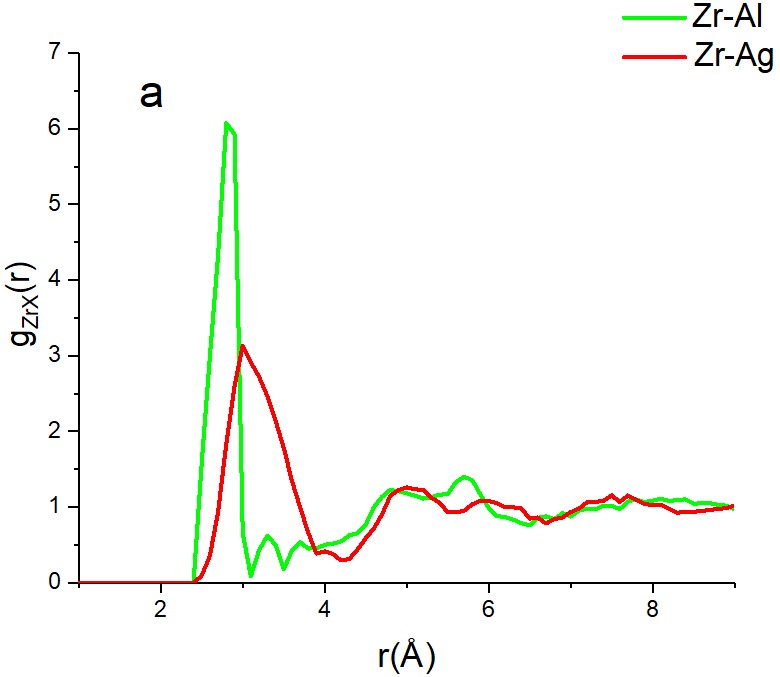
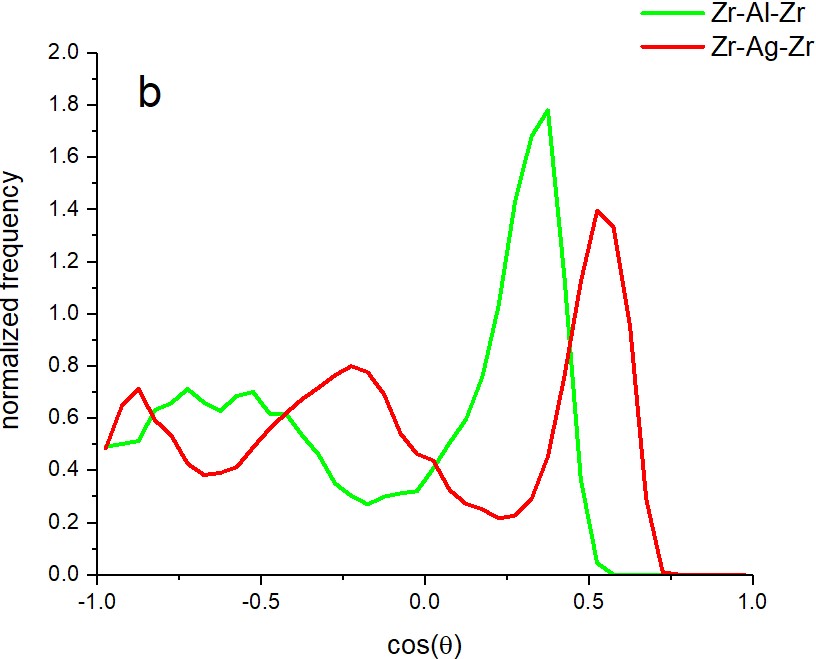
Figure 1. Effective size difference of Al and Ag atoms in the Cu-Zr host matrix as reflected by
the position of the first peak of Zr-Al and Zr-Ag partial pair distribution functions (a) and by
the Zr-Al-Zr and Zr-Ag-Zr cosine distributions (b)
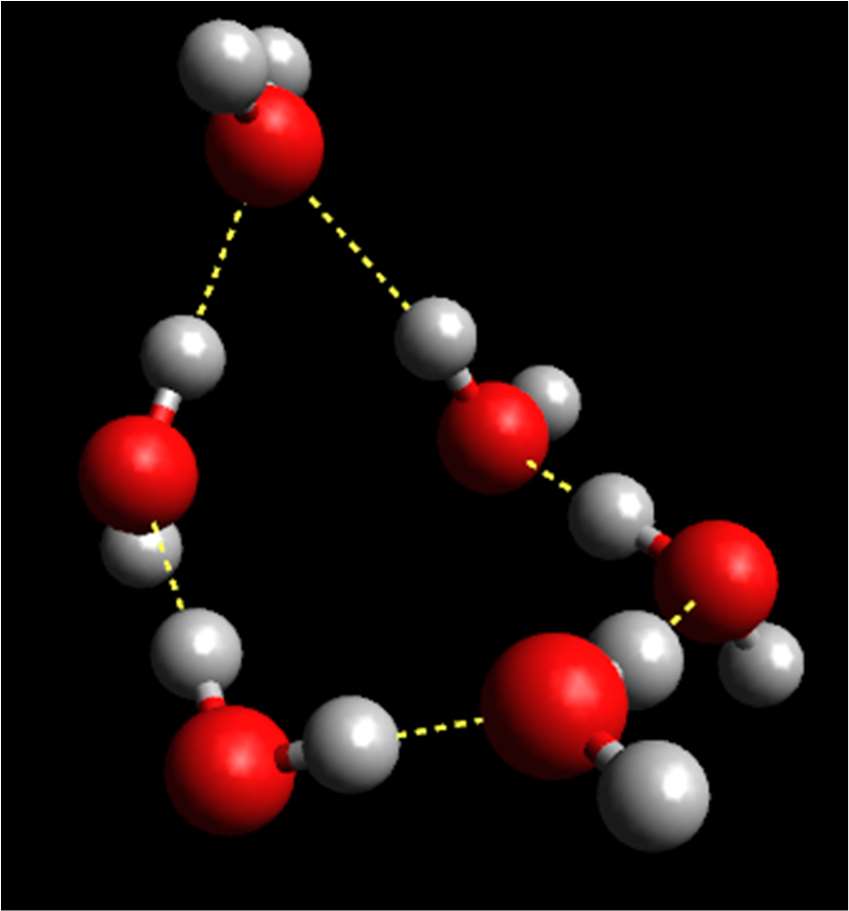
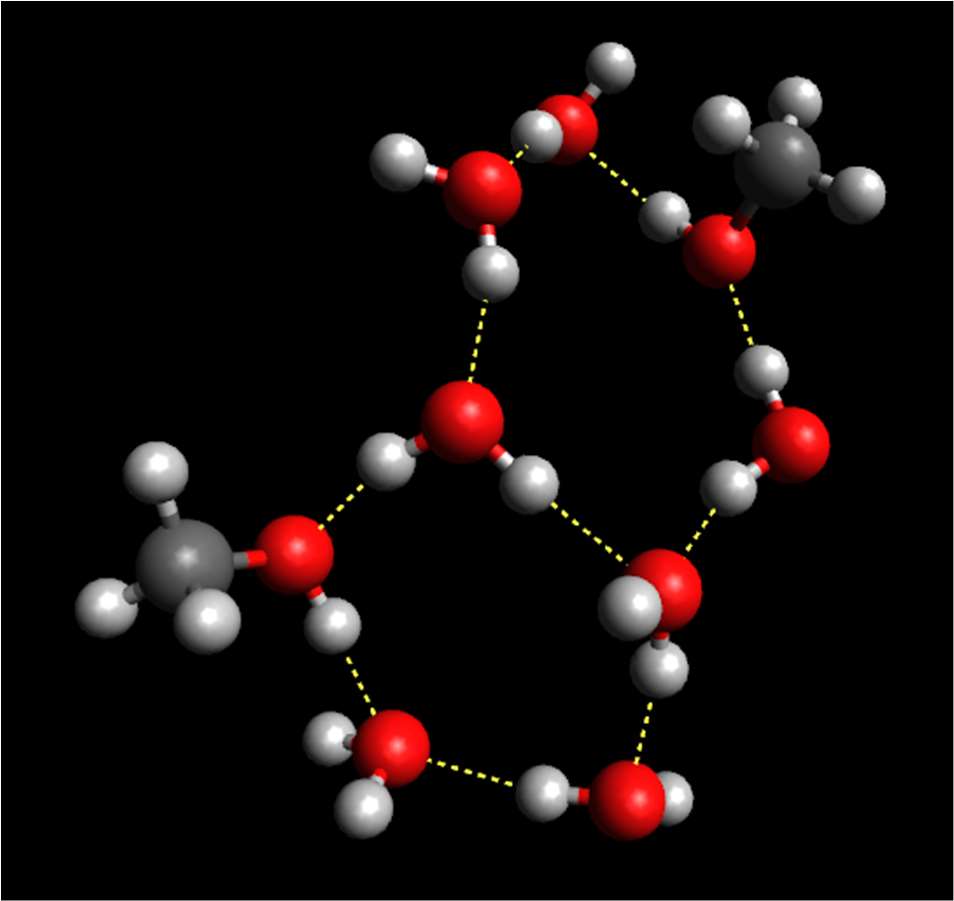
Figure 2. Hydrogen-bonded ring structures in methanol/water mixtures (Red: O atoms; light
grey: H atoms; dark grey: C atoms. Dashed lines represent hydrogen bonds between
molecules.)
Liquid chalcogenides. — The short-range order in the liquid state of GeTe, a prototypical
phase-change material employed in data storage devices, has been investigated by X-ray
and neutron diffraction in the temperature range from 1197 to 998 K. We have also
measured the dynamic viscosity from 1273 to 953 K, which is 55 K below the solidification
point, using an oscillating-cup viscometer. The measurements have been complemented
with ab-initio molecular dynamics (AIMD) simulations based on density functional theory
(DFT). Compatibility of the AIMD-DFT models with the diffraction data has been proven by
simultaneous fitting of all datasets in the frame of the reverse Monte-Carlo simulation
technique. It has been shown that octahedral order dominates in liquid GeTe, although
tetrahedral structures are also present. The temperature dependences of the structural
parameters, dynamic viscosity and electronic properties extracted from the AIMD models
have been analyzed and discussed. We have shown that GeTe keeps its semiconductor
nature in the liquid and supercooled liquid state. Its viscosity obeys the Arrhenius law with a
small activation energy of the order of 0.3 eV, which is indicative of a highly fragile liquid.
Aqueous salt solutions. — Aqueous lithium chloride solutions up to very high
concentrations have been investigated in classical molecular dynamics simulations. Various
force fields based on the 12-6 Lennard-Jones model parametrized for non-polarizable water
solvent molecules (SPC/E, TIP4P, TIP4PEw) have been inspected. Twenty-nine combinations
of ion-water interaction models have been examined at four different salt concentrations.
Densities, static dielectric constants and self-diffusion coefficients have been calculated
(see, e.g., Figure 3). Results derived
from the different force fields scatter
over a wide range of values. Neutron
and X-ray weighted structure factors
have also been calculated from the
radial distribution functions and
compared with experimental data. It
has been found that the agreement
between calculated and experimental
curves is rather poor for several
investigated potential models, even
though some of them were previously
applied in computer simulations.
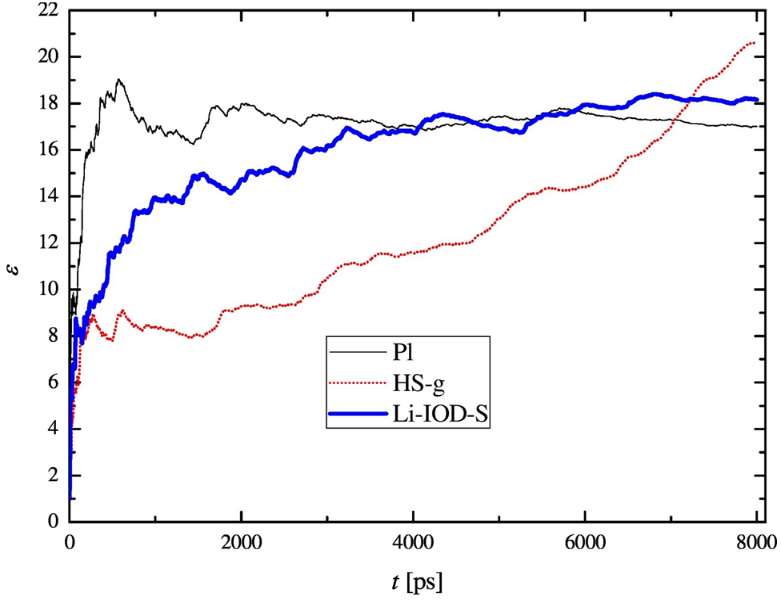
Figure 3. Convergence of the static dielectric constant (ε) for three selected models (Pl, HS-g
and Li-IOD-S) at the concentration m = 19.55 mol/kg. The curve is converged for the Pl
model, still slightly evolving for the Li-IOD-S model, and definitely not converged even at 8
ns, for the HS-g model.
Kapcsolat
Phone center: +36-1-392-2222
Secretary of the Director General: +36-1-392- 2512
Media contact: +36-30-487-9869